Physician Resources
Journals
- Nature Reviews Rheumatology (www.nature.com)
- Annals of the Rheumatic Diseases- (www.nature.com)
- Arthritis & Rheumatology- (www.onlinelibrary.wiley.com)
- Arthritis Research & Therapy- (www.arthritis-research.com)
- Current Opinion in Rheumatology- (www.journals.lww.com)
- Current Rheumatology Reports- (www.springer.com)
- Lupus- (www.lup.sagepub.com)
- Rheumatology- (www.rheumatology.oxfordjournals.org)
- The Journal of Rheumatology- (www.jrheum.org)
- Indian journal of rheumatology- (www.indianjrheumatol.com)
- Journal of Bone and Mineral Research- (www.jbmr.org)
- Arthritis Care & Research- (www.onlinelibrary.wiley.com)
- Seminars in Arthritis and Rheumatism- (www.semarthritisrheumatism.com )
- Best practice & research in clinical rheumatology- (www.elsevier.com)
- Clinical and Experimental Rheumatology- (www.clinexprheumatol.org)
- Rheumatic Disease Clinics of North America- (www.sciencedirect.com)
- Scandinavian Journal of Rheumatology- (www.tandfonline.com)
- Modern Rheumatology- (www.springer.com)
- BMC Musculoskeletal Disorders- (www.biomedcentral.com)
- Clinical Rheumatology- (www.springer.com)
- International Journal of Rheumatic Diseases- (www.wiley.com)
Classification Criteria
Dr Sundaram T G
SGPGI, Lucknow
Dr Avinash Jain
SGPGI, Lucknow
Vaccination in Autoimmune Rheumatic Disease
Prepared by
Dr Avinash Jain
SGPGI, Lucknow
Dr Rutviz Mistry
SGPGI, Lucknow
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma, Dr Praveen Hisaria and Dr Sapan C Pandya
Infections are the major cause of morbidity and mortality in patients with Autoimmune Rheumatic Diseases. Immune response in these patients is impaired and the “immunosuppressive” medications used to treat them add fuel to the fire. Infections are the biggest threat in the management of rheumatic conditions. Influenza, invasive pneumococcal infection, herpes zoster and hepatitis B are the major vaccine preventable infections seen in our patients.
Vaccination in rheumatic disease and its studies present with a unique set of challenges. Vaccination leads to immune response to particular antigen; however, non-specific response in this situation might lead to flare of the autoimmune disease. Ideally, studies on efficacy should use clinical endpoints to test the role of vaccines in rheumatic diseases to establish its clinical benefits. Such studies with clinical endpoints are logistically demanding and require a large sample size. Hence, most of the current studies use laboratory parameters (serologic titres of antibodies or T cell reactivity against antigen) to establish the efficacy of the vaccines. However, laboratory surrogates lack correlation with the clinical endpoints of reducing infection. Studies related to vaccination need to be interpreted with this consideration in mind.
In this write-up we will focus on the evidence of efficacy of various vaccines in rheumatic diseases and end with the current recommendations pertaining to vaccination.
Influenza virus
Influenza vaccine currently available in the market include inactivated and live attenuated. Trivalent and tetravalent vaccines containing three and four strains are available; however, the most commonly used is the trivalent inactivated vaccine. Multiple studies including a prospective1, and a retrospective large registry based Taiwanese study2 with clinical endpoints suggest reduction in pneumonitis, bronchitis, hospitalization in Rheumatoid arthritis and SLE patients vaccinated with influenza vaccine as compared to unvaccinated patients. In SLE, the serological response to the influenza subunit varied among different studies. Few studies show mild reduction in sero-protection while the others did not show any difference in seroconversionbetween vaccinated and unvaccinated patients.3,4 Serologic evidence of protection in Systemic Sclerosis5, Granulomatosis with polyangitis6 and Sjögren’s syndrome7 have been observed after influenza vaccination. Significant body of evidence exists to suggest efficacy of Influenza vaccination with concomitant use of glucocorticoids, csDMARDs and antiTNF therapy and tocilizumab. In one study, the arm on combination therapy with methotrexate and anti-TNF had lower titres of antibodies to influenza as compared to methotrexate alone, however, multiple other studies have shown good response with combination therapy as well.8 Studies with rituximab in RA however have documented significant lower seroconversion rates.9None of the studies have raised concerns regarding the safety or flare of underlying autoimmune disease.
Antigenic Drift and Shift leads to changing immunogenicity of the Influenza strains each year. Depending on the strains in circulation in a particular demographic area, the manufacturers “update” their vaccine to include the recent strains. This should be kept in mind while administering vaccine to patients. The best time to vaccinate with yearly shot is before the onset of monsoon10 (April-May) since influenza infection is particularly more common in monsoon and winter.
Thus, current body of evidence suggest influenza vaccines are well tolerated but underutilized in rheumatic diseases patients and are generally immunogenic even with immunosuppressants with the exception of rituximab.Vaccines should ideally be administered before B cell-depleting biological therapy [BCDT] is started or, when patients are on such a treatment already, at least 6 months after the start but 4 weeks before the next course.The European League Against Rheumatism (EULAR) recommend yearly vaccination with influenza of all patients with rheumatic diseases11.
Streptococcus pneumoniae
Currently two forms of pneumococcal vaccines are available. PPSV23 is derived from polysaccharide capsule while PCV13 is a conjugated vaccine with diphtheria carrier protein. PPSV23 response is T cell independent while PCV13 is T cell dependent. The immunological response is robust in PCV13 compared to PPSV23.Hence, boosters are required in PPSV23 while a single dose is sufficient in PCV13. Majority of the available literature has used PPSV23 in rheumatic diseases. Another reason for heterogeneity among the available data is lack of generally accepted serologic protection criteria for immunologic response to Pneumococcal vaccine.
In RA, good body of evidence exists to suggest adequate serologic response to pneumococcal vaccination independent of the DMARD used and disease activity12. However, newer studies have documented mildly reduced seroconversion with methotrexate-antiTNF combination and severely impaired humoral response with BCDT.13,14 Recent studies of PPSV23 in SLE suggest reduced immunogenicity as compared to healthy controls.15 Efficacy of pneumococcal vaccine is also established in Systemic sclerosis16 and Psoriatic arthritis.17
Center for Disease Control (CDC) recommends PCV-13 followed by PPSV-23 at least 8 weeks later for general population. For those who have already received PPSV-23, PCV-13 should be given at least 1 year later with and additional PPSV-23 booster given as usual 5 years from the first18
EULAR Guidelinesstrongly recommend pneumococcal vaccination inall patients with rheumatic diseases11.
Table 1: Immunogenicity of various vaccines in the presence of various immunosuppressants in RA and SLE
| Methotrexate | TNFi | Rituximab | Abatacept | Tofacitinib | Tocilizumab | |
|---|---|---|---|---|---|---|
| RA | ||||||
| Influenza | ± | + | ↓↓ | ↓ | + | + |
| Pneumococcal | +* | +* | ↓↓ | ↓ | ↓ | + |
| SLE | ||||||
| Influenza | + | + | ↓↓ | ↓ | NA | NA |
| Pneumococcal | ± | + | ↓↓ | NA | NA | NA |
* combination – reduced immunogenicity; ± Doubtful; ↓ Reduced; + Intact immunogenicity; NA,Not available; TNFi, TNF inhibitor
Hepatitis B
Studies in RA, SLE, Ankylosing Spondylitis, Behcet’s disease suggest immunogenicity of the Hepatitis B vaccination irrespective of disease activity, steroid or DMARD use. However, the amount of the data is insufficient to draw meaningful conclusions. EULAR guidelines recommend Hepatitis B vaccination for the patients at risk including intravenous drug abuse, multiple sex partners in the previous 6 months or health care personnel11. Hepatitis B vaccination is a part of universal immunization programme in India.
Herpes Zoster
Herpes zoster infection risk is increased in Rheumatic diseases. Special concerns regarding Herpes Zoster are being raised in view of increased risk in patients of RA receiving tofacitinib.As HZV is a live attenuated vaccine, its use in immunosuppressed patients is controversial. However, evidence is accumulating from larger registry based studies suggesting its safety in immunosuppressed patients with rheumatic diseases19. American Advisory Committee on Immunization Practices(ACIP) recommends using HZV in general population ≥ 50 years, persons anticipating immunosuppressant (at least two weeks prior to administration of immunosuppressive agent), in persons taking low-dose immunosuppressive therapy (e.g., <20 mg/day of prednisone or equivalent or using inhaled or topical steroids)20. Temporary discontinuation of immunosuppressive medication before vaccination with live attenuated vaccines might also be considered, but there are no studies to support this strategy.
Live vaccination should be avoided in following scenarios
- Steroids – steroid more than 10 mg for two weeks or more
- cDMARDs- Cyclosporine>2.5 mg/kg per day, Sulfasalazine >40 mg/kg per day or 2 g/day, Azathioprine>3 mg/kg,Cyclophosphamide >2.0 mg/kg per day, Leflumomide>0.5 mg/kg per day
- Biologic except B cell depletion therapy (BCDT) – Avoid anti TNF for four weeks
- BCDT – Avoid after BCDT for 6 months and can be given 4 weeks prior to BCDT initiation
Vaccine coverage in an outpatient rheumatology clinic in Germany were 18% and 25% for pneumococcal and influenza respectively.21 Another telephone based survey reported reasons for failure to receive pneumococcal and influenza vaccine were lack of doctor recommendations (55%), safety or efficacy concern (21%) and lack of motivation (19%)22. Simple interventions shown to be useful in increasing coverage include: presentation to rheumatology providers, creation of immunization algorithm, placing reminders on clinic forms, stocking the vaccine in clinic, establishing protocols for vaccination at admission.
To summarize, Box 2 shows the EULAR recommendations for the vaccination of individuals with AIRD. Recently, updates of these guidelines were presented in EULAR Meeting, Amsterdam, 2018.
- Vaccination status should be assessed in the initial work-up of patients
- Vaccine should ideally be administered to patients with an AIRD during stable disease
- Live attenuated vaccines should be avoided whenever possible.
- Vaccine can be administered to patients being treated with DMARDs and TNF inhibitors, but vaccine should be administered before starting B-cell-depleting biologic therapy
- Influenza vaccination should be strongly considered
- PPV23 should be considered
- Patients with an AIRD should have TT vaccination in accordance with the recommendations for the general population; in case of major or contaminated wounds in patients who received rituximab within 24 weeks, tetanus immunoglobulin instead of TT vaccine should be administered
- Herpes zoster vaccination “can” be considered
- For hyposplenic or asplenic patients, influenza, pneumococcal and H. influenzae type b and meningococcal C vaccinations are recommended
- HepatitisA and hepatitis B vaccination are only recommended for patients with an AIRD who are ‘at risk’ (i.e., intravenous drug abuse, multiple sex partners in the previous 6 months, or health care personnel)
- Patients who plan to travel are recommended to have vaccinations according to general rules, except for live-attenuated vaccines, which should be avoided whenever possible by immunosuppressed patients
- BCG vaccination is not recommended
It is under the process of publication. Newer recommendations include: immunocompetent household members of patients with AIIRD should be encouraged to receive vaccines according to national guidelines with the exception of oral poliomyelitis vaccine and live attenuated vaccine should be avoided for the first 6 months in newborn whose mother received biologics in second half of pregnancy.
bDMARDS and Vaccination
- Ideally, vaccination should be given (live or killed) four weeks before starting B cell depletion therapy. However, partial efficacy has been noted when given at least two weeks before Rituximab.
- Killed vaccine can be given during treatment with anti TNF, tocilizumab.
- JAK inhibitor predispose to Herpes Zoster reactivation. Herpes Zoster vaccine should be given at least two weeks before starting JAK inhibitor.
- Live attenuated vaccines should be avoided whenever possible.
Table 2 summarises immunogenicity of vaccines in various CTDs, disease flare and recommendations.
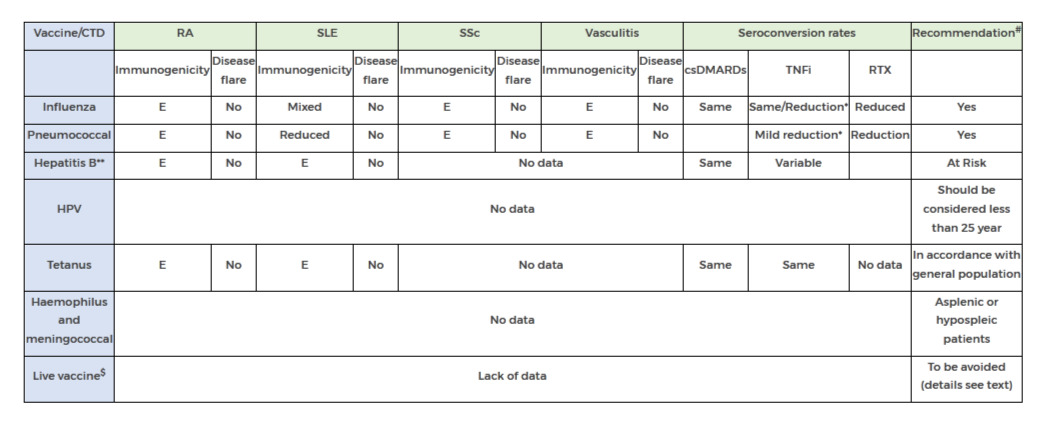
Table2: Efficacy of Vaccine in autoimmune rheumatic disease *with methotrexate; **more data needed; #see text for B cell depletion therapy; $BCG vaccine, oral poliomyelitis vaccine, oral typhoid fever vaccine and yellow fever vaccine csDMARDs, Conventional DMARDs; bDMARDs, biologics; E, Effective;RTX, Rituximab
REFERENCES
- 1. Stojanovich, L. Influenza vaccination of patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). Clin. Dev. Immunol.13, 373–375 (2006).
- 2. Chang, C.-C., Chang, Y.-S., Chen, W.-S., Chen, Y.-H. & Chen, J.-H. Effects of annual influenza vaccination on morbidity and mortality in patients with Systemic Lupus Erythematosus: A Nationwide Cohort Study. Sci. Rep.6, 37817 (2016)..
- 3. Louie, J. S. et al. Clinical and antibody responses after influenza immunization in systemic lupus erythematosus. Ann. Intern. Med.88, 790–2 (1978).
- 4. Holvast, A. et al. Safety and efficacy of influenza vaccination in systemic lupus erythematosus patients with quiescent disease. Ann. Rheum. Dis.65, 913–918 (2005).
- 5. Setti, M. et al. Flu vaccination with a virosomal vaccine does not affect clinical course and immunological parameters in scleroderma patients. Vaccine27, 3367–3372 (2009).
- 6. Holvast, A. et al. Wegener’s granulomatosis patients show an adequate antibody response to influenza vaccination. Ann. Rheum. Dis.68, 873–878 (2009).
- 7. Pasoto, S. G. et al. Short and long-term effects of pandemic unadjuvanted influenza A(H1N1)pdm09 vaccine on clinical manifestations and autoantibody profile in primary Sjögren’s syndrome. Vaccine31, 1793–1798 (2013).
- 8. Westra, J., Rondaan, C., Van Assen, S. & Bijl, M. Vaccination of patients with autoimmune inflammatory rheumatic diseases. Nat. Rev. Rheumatol.11, 135–145 (2015).
- 9. Kapetanovic, M. C. et al. Impact of anti-rheumatic treatment on immunogenicity of pandemic H1N1 influenza vaccine in patients with arthritis. Arthritis Res. Ther.16, R2 (2014).
- 10. Kumar, S., Rath, P. & Malaviya, A. A practical guide to adult vaccination for patients with autoimmune inflammatory rheumatic diseases in India. Indian J. Rheumatol.12, 160 (2017).
- 11. van Assen, S. et al. EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann. Rheum. Dis.70, 414–422 (2011).
- 12. Friedman, M. A. & Winthrop, K. Vaccinations for rheumatoid arthritis. Curr. Opin. Rheumatol.28, 330–336 (2016).
- 13. Bingham, C. O. et al. Immunization responses in rheumatoid arthritis patients treated with rituximab: Results from a controlled clinical trial. Arthritis Rheum.62, 64–74 (2010).
- 14. Kapetanovic, M. C. et al. Influence of methotrexate, TNF blockers and prednisolone on antibody responses to pneumococcal polysaccharide vaccine in patients with rheumatoid arthritis. Rheumatology45, 106–111 (2006).
- 15. Mathian, A., Pha, M. & Amoura, Z. Lupus and vaccinations. Curr. Opin. Rheumatol. 1 (2018). doi:10.1097/BOR.0000000000000525
- 16. MERCADO, U., ACOSTA, H. & DIAZ-MOLINA, R. Antibody Response to Pneumococcal Polysaccharide Vaccine in Systemic Sclerosis. J. Rheumatol.36, 1549–1550 (2009).
- 17. Mease, P. J. et al. Pneumococcal vaccine response in psoriatic arthritis patients during treatment with etanercept. J. Rheumatol.31, 1356–61 (2004).
- 18. Kobayashi, M. et al. Intervals Between PCV13 and PPSV23 Vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR. Morb. Mortal. Wkly. Rep.64, 944–947 (2015).
- 19. Yun, H. et al. Risk of Herpes Zoster in Autoimmune and Inflammatory Diseases: Implications for Vaccination. Arthritis Rheumatol.68, 2328–2337 (2016).
- 20. Dooling, K. L. et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR. Morb. Mortal. Wkly. Rep.67, 103–108 (2018).
- 21. Krasselt, M., Baerwald, C. & Seifert, O. Insufficient vaccination rates in patients with systemic lupus erythematosus in a German outpatient clinic. Z. Rheumatol.77, 727–734 (2018).
- 22. Lawson, E. F., Trupin, L., Yelin, E. H. & Yazdany, J. Reasons for failure to receive pneumococcal and influenza vaccinations among immunosuppressed patients with systemic lupus erythematosus. Semin. Arthritis Rheum.44, 666–71 (2015).
SAFETY AND USE OF MARS IN THE PRESENCE OF RENAL AND HEPATIC DYSFUNCTION
Authors
Dr Sakir Ahmed
KIMS, Bhubaneshwar
Ipsita Mohanty
Bhubaneshwar
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma
| DMARD | General Safety recommendations | Dose modificationin renal dysfunction. | Dose modification in liver dysfunction |
|---|---|---|---|
| Methotrexate | Monitor patients closely for bone marrow, liver, lung and kidney toxicities | CrCl 10-50 ml/min: 50% of dose at normal dosing interval CrCl<10 ml/min: avoid use | Bilirubin 3.1-5.0 mg/dl or AST> 3 times ULN: give 75% of dose Bilirubin >5.0 mg/dl: avoid use |
| Leflunomide | Can cause severe liver injury Recommend ALT monitoring monthly for 6 months after initiating, and q6-8weeks thereafter If ALT rises to >3x ULN, interrupt therapy while investigating probable cause; if likely leflunomide-induced, initiate cholestyramine washout to speed elimination and conduct follow-up LFTs at least weekly until ALT value within normal range; if not leflunomide-induced ALT elevation, may consider resuming leflunomide | There are no dosage adjustments provided in the manufacturer’s labelling | Not recommended for use in patients with pre-existing liver disease or those with baseline ALT>2 times ULN; monitor liver function closely. Use is contraindicated in hepatic impairment. |
| Sulfasalazine | Can lead to hepatobiliary disorders: reports of hepatotoxicity, including elevated liver function tests cholestatic jaundice, cirrhosis, hepatitis cholestatic, cholestasis and possible hepatocellular damage including liver necrosis and liver failure; Renal and urinary disorders: nephrolithiasis reported | Renal clearance: 37% There are no dosage adjustments provided in the manufacturer’s labelling; use with extreme caution | Data not available |
| Hydroxychloroquine | Both chloroquine and HCQ can cause a 10 percent decrease in creatinine clearance by competitively inhibiting creatinine secretion; this does not represent a true change in renal function. | Excretion of these drugs is principally by direct renal clearance of the parent compound and hepatic metabolites. Manufacture does not provide instructions for use in renal failure. Expert recommendation is reduction of dose <250mg/day. antimalarials have been found in the urine five years after medication was stopped | Data not available. Dose should be reduced if continued. |
| Azathioprine | Increased risk of infection and hepatotoxicity; monitor liver function periodically; hepatic sinusoidal obstruction syndrome reported; discontinue therapy if suspected | CrCl>50 ml/minute: no adjustment recommended. CrCl 10 to 50 ml/minute: administer 75% of normal dose. CrCl<10 ml/minute: administer 50% of normal dose. Haemodialysis (partially dialyzable; ~45% removed in 8 hours): administer 50% of normal dose; supplement: 0.25 mg/kg. CRRT: administer 75% of normal dose. | There are no dosage adjustments provided in the manufacturer’s labelling. However expert recommendation is that it may be used with caution. |
| Mycophenolate mofetil | Toxicity may increase in renal impairment; use caution | there have been no specific dosage adjustments identified, Although use of lower doses may be required. Mycophenolic acid (MPA) exposure appears to be inversely related to renal function. With GFR less than 25mL/min/1.73m2 in renal transplant recipients doses more than 2g/d should be avoided. | however, it is not currently known whether dosage adjustments are necessary for hepatic disease with other aetiologies. Increased monitoring may be necessary in patients with hyperbilirubinemia and/or hypoalbuminemia |
| Apremilast | Renal/hepatic impairment | Severe renal impairment (CrCl<30 ml/min): reduce dose to 30 mg po qday Mild-to-moderate renal impairment: no dosage adjustment required | Hepatic impairment: no dosage adjustment required |
| Tacrolimus | Increased mortality in female liver transplant patients. Renal impairment does not affect the elimination or serum concentrations of tacrolimus; however, tacrolimus may cause nephrotoxicity requiring dose reduction. Post-transplant hepatic impairment may be associated with increased risk of developing renal insufficiency | Use lower end of dosing range Monitor renal function and adjust dose according to whole blood concentrations and tolerability | Mild: no dosage adjustment required Moderate: monitor whole blood concentrations and adjust dose accordingly Severe (mean child-pugh score >10): mean clearance of tacrolimus was substantially lower compared with normal hepatic function; dosage reduction recommended; administer 80% of preconversion daily dose of immediate release dosage form when converting from tacrolimus immediate release to extended release |
| Cyclophosphamide | Use with caution in patients with hepatic or renal impairment | Renal impairment: CrCl<10 ml/min, give 75% of normal dose; CrCl>10 ml/min, give full dose | Hepatic impairment: give 75% of normal dose if transaminase levels are >3 times upper limit of normal or bilirubin is 3.1-5 mg/dl |
| Rituximab | Increased risk of potentially fatal hepatitis b virus reactivation | There are no dosage adjustments provided in the manufacturer’s labelling (has not been studied) | There are no dosage adjustments provided in the manufacturer’s labelling (has not been studied) |
| Infliximab Adalimumab Etanercept Golimumab | There are no dosage adjustments provided in the manufacturer’s labelling. There are case reports of successful use in renal or hepatic failure. | ||
| Secukinumab | Data not available | Data not available | Data not available |
| Tofacitinib | Associated with increased LFTs | Mild: no dosage adjustment required RA or PsA Moderate-to-severe: not to exceed 5 mg qday UC Moderate-to-severe: if taking 10 mg bid, reduce to 5 mg bid; if taking 5 mg bid, reduce to 5 mg qday | Mild: no dosage adjustment required Severe: not recommended Ra or PsA Moderate-to-severe : not to exceed 5 mg qday UC Moderate-to-severe: if taking 10 mg bid, reduce to 5 mg bid; if taking 5 mg bid, reduce to 5 mg qday |
| Baricitinib | Data not available | Renal impairment Egfr ≥60 ml/min/1.73 m²: renal function significantly affects Baricitinib systemic exposure; monitor closely eGFR<60 ml/min/1.73 m²: not recommended | Hepatic impairment Mild or moderate: no dose adjustment required Severe: not recommended |
Pregnancy and Rheumatic Disease
Authors
Dr Sakir Ahmed
KIMS, Bhubaneshwar
Ipsita Mohanty
Bhubaneshwar
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma
Reviewed and Edited by : Dr Avinash Jain
This document is a summary of guidelines of EULAR, ACR and BSR along with the inputs from a literature search. It is designed to be brief and in simple language understandable both to the patient and the specialist. However patients are strongly advised not to interpret the drug advisory on their own without consulting their treating physicians.
A. A successful pregnancy is possible in almost all rheumatic diseases provided disease is well-controlled, and there is no permanent organ damage.
- 1. Pregnancies are more likely to be successful when they are planned, with adequate discussion among the patient, the rheumatologist and the obstetrician.
- 2. Successful, however, does not mean uneventful. Doctors and patients must be prepared to deal with possible complications for both mother and child.
- 3. Diseases with the potential to affect the kidneys, or lung (including increasing pressure of the pulmonary arteries) like lupus, antiphospholipid syndrome, inflammatory myositis, systemic sclerosis and overlap syndromes are more likely to affect pregnancy outcome than others.
- 4. Persistently raised creatinine (end-stage renal disease) or high pulmonary arterial pressures may hinder successful pregnancies. In fact in these conditions, pregnancy may worsen the health condition of the mother.
- 5. Any rheumatic diseasemust be under optimal control for 6 months before pregnancy is planned.
B. Effects of rheumatic diseases on pregnancies:
- 1. In the absence of permanent organ damage, the fertility of patients is not altered due to rheumatic diseases. Diseases like systemic sclerosis or sjogrensyndrome may lead to dyspareunia.
- 2. Uncontrolled rheumatic diseases have a lot of inflammation and may lead to pregnancy loss especially in the 1st
- 3. In the later phase there are more chances of pregnancy induced hypertension and foetal growth retardation. Patients who have or have had kidney disease, due to vasculitis, scleroderma, or lupus, generally have an increased risk of severe hypertension and pre-eclampsia.
- 4. Pulmonary arterial hypertension worsens in the post-partumperiod.Patients with high pulmonary artery pressures are advised not to get pregnant.
- 5. APS probably has the greatest impact on pregnancy. It causes both early and late miscarriage.Other complications include premature birth, low-weight babies, athrombosis (condition where blood clots form in the blood vessels) and pre-eclampsia. Thus, pregnancy with APS should always be considered as high risk and require close medical and obstetric monitoring. Treatment is based on low-dose aspirin and heparin.
- 6. Babies of mothers having anti-Ro antibodies (in Sjogren or lupus) are at higher risk of developing congenital heart blocks. The anti-Ro antibodies may interfere with the development of the electric conduction system of the heart. Thus mother with anti-Ro antibodies needsfoetal heart monitoring with foetal echocardiography (ultrasound of foetalheart) during 2nd
- 7. It is important to discuss the possible effects of various anti-rheumatic drugs on pregnancy. There should be expert assessment of risk-benefit to determine the drugs to be continued during pregnancy starting from the pre-conception stage.
C. Effect of pregnancies on rheumatic diseases:
- The earlier paradigm was that diseases like rheumatoid arthritis tend to go into quiescence (2/3rd of RA) while diseases like lupus would invariably flare (50% of SLE flare with 20% being major organ flares) during pregnancy.
- With newer treatment strategies and better disease control, studies have shown that only a minority (~10%) of RA have improved disease activity while lupus patients (in remission for at least 6 months, and on hydroxychloroquine) do not have increased flare rates.
- It is very important to report any new symptoms and any worsening to both your rheumatologist and obstetrician.
D. Drugs permissible in pregnancy:
- Previously drugs were prescribed followingUSFDA pregnancy categories: A, B, C, D, and X. However these are not water-tight compartments and this has lead the FDA to abandonthis approach. Thus, risk-benefit of each drugs should be discussed in context of the patient, the disease and the age of gestation.
- As overarching guidelines, we endorse the ACR recommendation that currently stand as: (please also see the BSR guidelines: table 2)
| Pregnancy | Lactation | ||
|---|---|---|---|
| NSAID | Yes (avoid after 32 weeks) | Yes | |
| Sulfasalazine | Yes | Yes | |
| Antimalarials | Yes | Yes | |
| Corticosteroids | Yes | Yes | |
| Cyclosporine/Tacrolimus | Yes | Probably yes | |
| Azathioprine | Yes | Probably yes | |
| Mycophenolate | No | No | |
| Methotrexate | No | No | |
| Cyclophosphamide | No | No | |
| Anti-tumor necrosis factor (TNF) | Yes | Yes | |
| Rituximab | No | No | |
| Warfarin | No (with caution, only after first trimester) | Yes | |
| Heparin | Yes | Yes | |
| Table 1: Acceptable medications during pregnancy and lactation | |||
- There is strong evidence that hydroxychloroquine must be continued in lupus and APS during pregnancy and it is our personal opinion that it should be continued in RA during pregnancy as well.
- Captopril and enalapril are to be avoided in pregnancy but are safe drugs during breastfeeding.
- Other drugs:
Tocilizumab
- Tocilizumab (TCZ) should be stopped at least 3 months before conception, but unintentional exposure early in the first trimester is unlikely to be harmful
- There are no data upon TCZ use in breastfeeding
Abatacept
- There are insufficient data to recommend abatacept (ABA) in pregnancy. Unintentional exposure early in the first trimester is unlikely to be harmful
- There are no data upon ABA use in breastfeeding
Secukinumab
- There are insufficient data to recommend Secukinumab(SEK) in pregnancy.
- There are no data upon SEK use in breastfeeding
Preconception planning for males:
- Limited data is available but it has shown that Methotrexate is unlikely to affect male fertility (table 2). Currently there are no recommendations to stop it during pre-conception stage.
- For sulfasalazine, conception rates may be enhanced by stopping sulfasalazine for 3 months prior to conception
- Thalidomide should be stopped at least 3 months in advance prior to planned conception (as it is present in spermatozoa).
Summary::
- The likelihood of a successful and healthy pregnancy is highest if kidney and heart function, and blood pressure are normal.
- The disease is inactive for at least 6 months prior to conception.
- Drugs should be used after patient tailored risk-benefit assessment.
Table 2: BSR guideline for drugs used in rheumatology during pregnancy:
| Summary of drug compatibility in pregnancy and breastfeeding | |||||
|---|---|---|---|---|---|
| Compatible peri-conception | Compatible with first trimester | Compatible with second/third trimester | Compatible with breastfeeding | Compatible with paternal exposure | |
| CORTICOSTEROIDS | |||||
| Prednisolone | Yes | Yes | Yes | Yes | Yes |
| Methylprednisolone | Yes | Yes | Yes | Yes | Yes |
| ANTIMALARIALS | |||||
| Hydroxychloroquine | Yes | Yes | Yes | Yes | Yes* |
| ANTIMALARIALS | |||||
| DMARDS | |||||
| Methotrexate <20mg/week | Stop 3 months in advance | No | No | No | Yes* |
| Sulfasalazine (with 5mg folic acid) | Yes | Yes | Yes | Yes† | Yes† |
| Leflunomide | Cholestyramine washout, no | No | No | No Data | Yes* |
| Azathioprine <2mg/kg/day | Yes | Yes | Yes | Yes | Yes |
| Ciclosporin | Yes | Yes§ | Yes§ | Yes* | Yes* |
| Tacrolimus | Yes | Yes§ | Yes§ | Yes* | Yes* |
| Cyclophosphamide | No | No| | No| | No | No |
| Mycophenolate mofetil | Stop 6 weeks in advance | No | No | No | Yes* |
| Intravenous immunoglobulin | Yes | Yes| | Yes | Yes | Yes* |
| ANTI-TNF | |||||
| Infliximab | Yes | Yes | Stop at 16 weeks | Yes* | Yes* |
| Etanercept | Yes | Yes | Second but not third | Yes* | Yes* |
| Adalimumab | Yes | Yes | Second but not third | Yes* | Yes* |
| Certolizumab | Yes | Yes | Yes* | Yes* | No data |
| Golimumab | No data | No data | No data | No data | No data |
| Cyclophosphamide | No | No| | No| | No | No |
| Cyclophosphamide | No | No| | No| | No | No |
| OTHER BIOLOGICS | |||||
| Rituximab | Stop 6 months in advance | No¶ | No | No data | Yes* |
| Tocilizumab | Stop 3 months in advance | No¶ | No | No data | No data** |
| Anakinra | No | No¶ | No | No data | No data** |
| Abatacept | No | No¶ | No | No data | No data** |
| Belimumab | No | No¶ | No | No data | No data** |
| CONVENTIONAL PAINKILLERS | |||||
| Paracetamol | Yes | Yes†† | Yes†† | Yes | Yes‡‡ |
| Codeine | Yes | Yes | Yes | Caution | Yes‡‡ |
| Tramadol | Yes | Yes | Yes | Yes§§ | Yes‡‡ |
| OTHER CHRONIC PAIN TREATMENTS | |||||
| Amitriptyline | Yes | Yes | Yes | Yes | Yes‡‡ |
| Gabapentin | No | Insufficient data|| | Insufficient data|| | Insufficient data | No data |
| Pregabalin | No data | No data | No data | No data | No data |
| Venlafaxine | Yes | Yes | Yes | Insufficient data|| | Yes‡‡ |
| Fluoxetine | Yes | Yes | Yes | Caution|| | Yes‡‡ |
| Paroxetine | Yes | Yes | Yes | ICaution|| | Yes‡‡ |
| Sertraline | Yes | Yes | Yes | ICaution|| | Yes‡‡ |
| NSAIDS | |||||
| NSAIDs | Yes | Caution¶¶ | Stop by week 32 | Yes | Yes |
| COX-2 inhibitors | No | No | No | No | No data |
| Low-dose aspirin | Yes | Yes | Yes | Yes*** | Yes‡‡ |
| ANTICOAGULANTS | |||||
| Warfarin | No | No | No/Caution | Yes | No data |
| Low-molecular-weight heparin | Yes | Yes | Yes | Yes*** | Yes‡‡ |
| Dabigatran | No data | No data | No data | No data | No data |
| BISPHOSPHONATES | |||||
| Bisphosphonates | Stop 6 months in advance | No | No | No data | No data |
| ANTIHYPERTENSIVES | |||||
| Angiotensin-converting-enzyme inhibitor | Stop when pregnancy confirmed | No | Yes§§ | No data | |
| Nifedipine | Yes | Yes<60mg/day | Yes<60mg/day | Yes | Yes‡‡ |
| Amlodipine | No data | No data | No data | No data | Yes*** |
| PULMONARY VASODILATORS | |||||
| Sildenafil | No data | No data | No data | No data | No data |
| Bosentan | No data | No data | No data | No data | No data |
| Prostacyclin | No data | No data | No data | No data | No data |
| NSAIDS=non-steroidal anti-inflammatory drugs; COX-2=cyclooxygenase-2; MDT=multidisciplinary team. * Data are limited † In healthy full-term infants only ‡ Conception may be enhanced by stopping sulfasalazine for 3 months prior to conception § Suggested monitoring of maternal blood pressure, renal function, blood glucose and drug levels | Only consider in severe or life-/organ-threatening maternal disease ¶ Unintentional first trimester exposure is unlikely to be harmful ** Unlikely to be harmful †† Intermittent use advised, see full guideline for details ‡‡ No studies identified, but unlikely to be harmful due to maternal compatibility §§ Limited evidence, but unlikely to be harmful || Insufficient evidence regarding use for treatment of chronic pain in pregnancy ¶¶ Possible association with miscarriage and malformation *** No studies identified, but unlikely to be harmful. | |||||
Table 3 The impact of rheumatic disease on pregnancy and vice-versa.
| Rheumatic Diseases | Impact of pregnancy on rheumatic disease | Pregnancy outcome > IUGR/premature/SGA | Foetal Loss | Other complications | Postpartum | Fertility |
|---|---|---|---|---|---|---|
| RA | Decreased disease activity (found in only a minority of recent studies under T2T regimens) | Patients on glucocorticoids maybeat risk for small for gestational age and for preterm delivery | not been convincingly shown to be associated with an increase in foetal morbidity or foetal losses | pregnancy outcomes in women with well-controlled RA are comparable to those in the general population | Flares in up to 90%.Usually not in well controlled disease. | Preserved. |
| SLE | Increased flare rates. Patients on HCQ have similar flare rates as non-pregnant counterparts | extremely variable rate of induced abortions reported. Possibility of CHB if anti-Ro present in mother. Concomitant APS increases risk | ~5% | two- to fourfold increased rate of obstetric complications including preterm labour, unplanned caesarean delivery, foetal growth restriction, preeclampsia, and eclampsia. Patients with SLE also have significantly higher risk of thrombosis, infection, thrombocytopenia, and transfusion | Flares. Usually not in well controlled disease. | Preserved in well controlled lupus without organ damage. High dose CYC is a risk factor for reduced fertility. Apparent infertility is common due to effects on their own self-esteem and mental well-being, and stress with partner. |
| APS | Potentially increased risks of thrombosis especially in post-partum period | Up to 50% of treated cases have PIH and related foetal complications | up to 80% risk of current pregnancy loss without treatment. 20% with treatment | Operative deliveries are commoner; increased risk of PIH, placental insufficiency and abruption, HELLP syndrome and pre-term labour. Rarely foetal thrombosis | Increased risk of thrombosis | Much lowered |
| SSc | Less data available. May exacerbate vasculopathy like PAH, raynaud, or risk for renal crisis | Limited data: possibly more premature births and more infants small for gestational age | Multiple studies (but not all) suggest increased risk of abortion; but most have small numbers | increased frequency of preterm delivery, intrauterine growth restriction, and low-birthweight baby | Data is less but apparently fertility is maintained. Dyspareunia can be an issue | |
| Sjogren | Like lupus: likely to worsen during pregnancy and more so in the postpartum period, especially in presence of PAH | Increased risk of foetal growth restriction. | variable rate of induced abortions | prevalence of CHB is 1-2%. Recurrence rates are 10-20%. Neonatal lupus risk is ~2% | Flares. Flares expected to be less in well controlled disease | Data is less but apparently fertility is maintained in absence of organ damage. Dyspareunia can be an issue |
| Takayasu | Unknown; theoretically possible to increase long term morbidity | ~25% have growth retardation; | 25% | Operative deliveries are commoner (~40%); increased risk of PIH and pre-term labour | Unknown | Data is less. Major determinants of fertility are hypertension, cardiac involvement and renal (artery) involvement |
| ANCA associated vasculitis | Data is very limited but around 20% flare during pregnancy | Limited data | 10% of cases in GPA, up to 20% in EGPA (under optimal conditions) | 20% preterm | Unknown | Decreased |
T2T: treat to target strategy; PAH: pulmonary arterial hypertension; HCQ: hydroxychloroquine; ANCA: anti-neutrophil cytoplasmic antibodies; CHB: complete heart block
Osteoporosis and Rheumatic Disease
Author
Dr Avinash Jain
SGPGI, Lucknow
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma, Dr Praveen Hisaria and Dr Sapan C Pandya
This is an area which is often neglected and is a burden too heavy to be carried by the weakened bones! There is an increased risk of osteoporosis (OP) in rheumatic diseases, a chronic inflammatory state, often warranting the use of steroids. Pathogenesis is multifactorial involving cross-talk between inflammatory cells and bone cells, disease complications, poor nutrition, medications, and decreased physical activity. Dickkopf-related protein 1 (DKK-1) and sclerostin, which are negative regulators of the Wntsignalling pathway, inhibit bone formation in rheumatic diseases.[1] Steroid use increases their expression besides augmenting osteoclastogenesis by inhibiting Osteoprotegerin (OPG) and increasing RANKL expression. Muscle wasting and changes in bone microstructure further compound the problem. Factors which increase the risk of OP have been outlined in Box
1.OP screening involves
- 1. A good history and examination to asses risk factors and h/o fracture and examination including BMI and loss of height, reduced space between lower ribs and pelvis, spinal tenderness.
- 2. Look for secondary causes
- 3. Addictions particularly tobacco use
- 4. Medications
- 5. Biochemistry including calcium, ALP
- 6. Bone mineral density (BMD) assessment within six months of initiation of glucocorticoid treatment. Most widely used tool is Dual-energy x-ray absorptiometry (DXA) thought newer techniques like pDXA (peripheral DXA), Quantitative Computerised Tomography (QCT) and peripheral QCT are available but needs validation and are costlier.
- 7. Biochemical biomarkers like C telopeptide, free deoxypyridinoline may independently predict fracture risk but are not routinely used
Box 1: Risk Factors
- 1. Age
- 2. Previous h/o low trauma fractures
- 3. Low BMI, Significant weight loss
- 4. Current smoking and alcohol
- 5. Parental h/o hip fracture
- 6. Use of steroids
- 7. Rheumatic diseases
- 8. Secondary OP – Endocrine causes, IBD, malabsorption, Chronic liver disease
- 9. Malnutrition
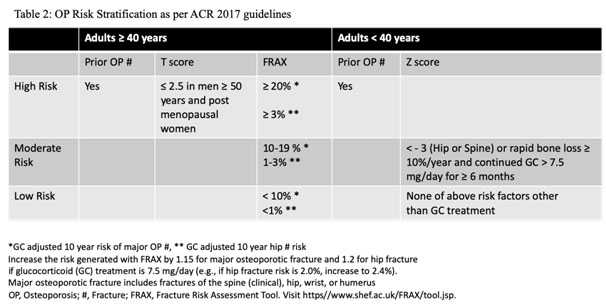
Fracture Risk Assessment Tool (FRAX) developed by University of Sheffield, estimates 10- year probability of hip and major OP fracture (hip, clinical spine, proximal humerus, or forearm) between 40-90 years using clinical risk factors with/without femoral neck BMD.[2] Besides age, gender, weight, height, it includes risk factors as defined in Box 1 where only Rheumatoid Arthritis is taken in rheumatic diseases, steroid use has been defined as ≥ 5mg for 3 or more months. There are however insufficient data to develop prediction tools for younger adults and children.
Although the greatest relative risk of fracture is in individuals with osteoporosis(OP), the absolute number of fractures in those with BMD T-scores in the low bone mass (osteopenia) range is the same or greater than in those with T-scores in the osteoporosis range, as more individuals belong to the latter category. At times BMD may give us a fall sense of hope and some patients who need preventive therapy may be missed.
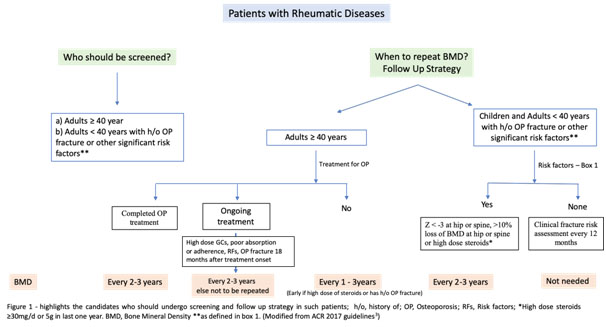
Rheumatic Diseases
Various studies have lookedat the prevalence of osteoporosis in rheumatic disease. [4,5] Prevalence and some of the associated risk factors have been mentioned in Table 1.
Table 1: Prevalence and Risk Factors of OP in rheumatic diseases[4,5]
| Osteoporosis | Additional Risk Factors | |
|---|---|---|
| SLE | 1.4 – 68% | Reduced sun exposure, high falls, renal involvement, longer disease duration No relation to disease activity |
| RA | 18 – 56% | Longer disease duration, Disease activity, HAQ, RF/ACPA, High CRP |
| AS | 19 – 62% | Older age, long-standing disease, syndesmophyte formation, associated IBD |
| PsA | 11 – 47% | Association with disease duration is controversial |
| SSc | 3 – 51% | Intestinal malabsorption, renal disease, subcutaneous calcinosis, no difference in lcSSc and dcSSc |
| IIM | 25% | Correlation with disease activity unclear |
| JIA | 40-52% of adult patients with JIA | Disease Activity and duration |
It is important to remember that osteoarthritis, syndesmophytes, new bone formation, atherosclerosis may falsely increase BMDmeasurement.[6,7]Management strategy should include FRAX score calculation and risk stratification and the use of OP medications accordingly. Moderate to highrisk patients should be treated with OP medications even if steroids are not used. (Table 2 and Figure 1)
GIOP (Glucocorticoid induced Osteoporosis)
Long-term glucocorticoid therapy causes osteoporotic fractures in about 10-12% of treated adult patients and 30–40% of them have radiographic evidence of vertebral fractures,
in view of higher effects of steroids on trabecular bone[8,9]Daily doses of ≤5 mg prednisolone have been shown to increase fracture risk by ~20%, rising to 60% for doses of ≥20 mg per day.[7,10]Fracture risk is highest particularly within first the 3-6 months and correlates with cumulative dose and daily dose. [10] Still the preventive care is suboptimal and less than a quarter undergo OP assessment and often the therapy is instituted once a fracture occurs.
Whom to Treat?
In GIOP, the terms ‘prevention’ and ‘treatment’ distinguish between the initiation of anti-osteoporosis intervention at the start of glucocorticoid therapy or after >3 months, respectively.
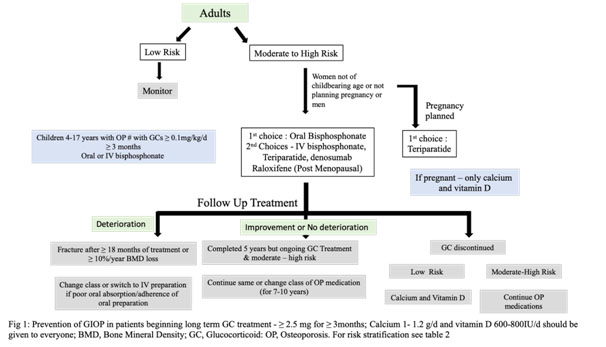
Prevention:
Depending on risk stratification, all adults irrespective of age, not of childbearing age or childbearing age but not planning a pregnancy during treatment, with moderate to high risk as defined in Table 2, should receive oral Bisphosphonate. Second line therapies include IV bisphosphonate, teriparatide, denosumab and raloxifene(for post-menopausal women only) in that order except in women of childbearing potential where teriparatide is preferred over oral bisphosphonate. IV bisphosphonate and denosumab lack safety data in pregnancy and hence should be used only in high risk patients whereas raloxifeneis not recommended. Some authorities do no rate bisphosphonates over teriparatide due to lack of head to head comparisons. Denosumab, though not a first line therapy, but is a good alternative particularly in patients with renal failure. Some experts recommend upfront teriparatide in patients with T-score < -3.5 or T-score of -2.5 or below plus a fragility fracture.Romosozumab, sclerostin inhibitor, is another new emerging therapy yet to find a place in guidelines.Refer to table 3 for dosing, side effects and monitoring.
Otheressential recommendations including for those with low risk of fracture are
- 1. Dailycalcium and vitamin D 1,000–1,200 mg and 600–800 IU respectively
- 2. Lifestyle modifications like cessation of smoking, limiting alcohol intake to 1-2/day,
- 3. Balanced diet and weight and daily exercises.
- 4. Figure 1summarises the prevention strategy and follow up treatment[11]In case of moderate to high risk, continue oral Bisphosphonate for ten years and Zoledronic acid for five years. There is no concept of drug holiday in such cases. Besides GCs, Methotrexate, Cyclophosphamide, heparin, oral anticoagulants, anti-convulsant are some of the medications which have been implicated in OP
Table 2
| Drug | Dose | Pre-requisites or monitoring | Side effects and Contra-indications |
|---|---|---|---|
| Oral Bisphosphonate | Alendronate 70mg/week Risedronate 35mg/week or 150mg/month | Avoid oral calcium supplements, antacids, magnesium supplements/laxatives, and iron preparations for at least 60 minutes after drug administration Check serum calcium | GERD/Reflux oesophagitis Osteonecrosis of Jaw (ONJ) – 1 in 10,000 to 1 in 100,000 patient-years Atypical Femur Fracture(AFF) Hypersensitivity GFR < 30ml/min Hypocalcemia achalasia, esophageal stricture, esophageal varices, Barrett’s esophagus) or with an inability to follow the dosing requirements (eg, stay upright for at least 30 minutes) Pregnancy and breastfeeding |
| IV Bisphosphonate | Zoledronic 4mg every year | Can have flu like symptoms, give Paracetamol before IV administration Check calcium and correct before drug administration | Transient hypocalcemia, Flu like symptoms within 24-72 hours of infusion; Treat with Paracetamol or NSAIDs ONJ, AFF Hypersensitivity GFR < 30ml/min Hypocalcemia Pregnancy and breastfeeding |
| Teriparatide | 20 mcg s.c every day upto 2 years | Hypercalcemia, Dizziness, arthralgia, rhinitis Osteosarcoma Hypersensitivity, hypercalcemia and related conditions | |
| Denosumab | 60mg s.c every six months | Check calcium and vitamin D and in deficient – correct same before administering Denosumab months Check calcium and vitamin D and in deficient – correct same before administering Denosumab Higher risk in CKD pts, eGFR < 30 and in those with malabsorption and hence more prone to hypocalcemia. Check calcium level 10 days after administration of Denosumab | Infections Skin Rash Immediate risk of fractures on stopping the drug – add bisphosphonate if denosumab needs to be stopped Hypersensitivity Hypocalcemia |
| Oral Bisphosphonate | Alendronate 70mg/week Risedronate 35mg/week or 150mg/month | void oral calcium supplements, antacids, magnesium supplements/laxatives, and iron preparations for at least 60 minutes after drug administration Check serum calcium | GERD/Reflux oesophagitis Osteonecrosis of Jaw (ONJ) – 1 in 10,000 to 1 in 100,000 patient-years Atypical Femur Fracture(AFF) Hypersensitivity GFR < 30ml/min Hypocalcemia achalasia, esophageal stricture, esophageal varices, Barrett’s esophagus) or with an inability to follow the dosing requirements (eg, stay upright for at least 30 minutes) Pregnancy and breastfeeding |
| IV Bisphosphonate | Zoledronic 4mg every year | Can have flu like symptoms, give Paracetamol before IV administration Check calcium and correct before drug administration | Transient hypocalcemia, Flu like symptoms within 24-72 hours of infusion; Treat with Paracetamol or NSAIDs ONJ, AFF Hypersensitivity GFR < 30ml/min Hypocalcemia Pregnancy and breastfeeding |
| Teriparatide | 20 mcg s.c every day upto 2 years | Hypercalcemia, Dizziness, arthralgia, rhinitis Osteosarcoma Hypersensitivity, hypercalcemia and related conditions | |
| Denosumab | 60mg s.c every six months | Check calcium and vitamin D and in deficient – correct same before administering Denosumab Higher risk in CKD pts, eGFR < 30 and in those with malabsorption and hence more prone to hypocalcemia. Check calcium level 10 days after administration of Denosumab | Infections Skin Rash Immediate risk of fractures on stopping the drug – add bisphosphonate if denosumab needs to be stopped Hypersensitivity Hypocalcemia |
CKD, Chronic Kidney Disease; GERD, Gastro-esophageal reflux disease; GFR, Glomerular filtration rate;s.c, subcutaneous
Osteoporosis is a potentially preventable complication which needs to be taken care of by a rheumatologist considering an increasing prevalence of OP in rheumatic diseases. Good control of disease and hence inflammation, minimising steroid use, emphasis on dietary and lifestyle modifications, calcium and vitamin D supplementation and use of OP medications when indicated will help in reducing this complication. “Prevention is always better than cure!Action is better than procrastination.”
Summary
Osteoporosis is a common but neglected issue, amenable to therapy or prevention in patients with rheumatic diseases particularly on steroids
- 1. 10-12% patients on steroid suffer from fracture or 30-50% have radiological evidence of vertebral fracture with maximum loss occurring in 3-6 months.
- 2. Do not forget secondary causes which further increase the risk of osteoporosis.
- 3. All patients with rheumatic diseases, age 40 years or more, and children with risk factors should undergo BMD within six months of initiation of glucocorticoid.
- 4. FRAX score, with correction for steroid dose, can be used in patients aged³ 40 years for fracture risk prediction with/without BMD. (https//www.shef.ac.uk/FRAX/tool.jsp)
- 5. Treatment should be started for those falling in moderate to high risk.
- 6. There is no role of follow up BMD adults population on treatment unless there are risk factors and/or development of fracture despite ³ 18 months of therapy or poor adherence/compliance.
- 7. There is no concept of drug holiday at end of 3-5 years if there is moderate to high risk and therapy should be continued for additional 3-5 years.
- 8. Upfront Teriparatide maybe considered in T-score < -3.5 or T-score of -2.5 or below plus a fragility fracture or a contra-indication for bisphosphonate except pregnancy
References
- 1. Rizzoli R, Biver E. Glucocorticoid-induced osteoporosis: Who to treat with what agent? Nat Rev Rheumatol 2015;11(2):98–109.
- 2. Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008;19(4):385–97.
- 3. Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Rheumatol 2017;69(8):1521–37.
- 4. Maruotti N, Corrado A, Cantatore FP. Osteoporosis and rheumatic diseases. Reumatismo 2014;66(2):125
- 5. Gao L-X. Osteoporosis in rheumatic diseases. World J Rheumatol 2015;5(1):14–23.
- 6. van der Weijden MAC, Claushuis TAM, Nazari T, Lems WF, Dijkmans BAC, van der Horst-Bruinsma IE. High prevalence of low bone mineral density in patients within 10 years of onset of ankylosing spondylitis: a systematic review. Clin Rheumatol 2012;31(11):1529–35.
- 7. Staa TP Van, Geusens P, Bijlsma JWJ, Leufkens HGM, Cooper C. Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum 2006;54(10):3104–12.
- 8. Curtis JR, Westfall AO, Allison J, Bijlsma JW, Freeman A, George V, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum 2006;55(3):420–6.
- 9. Angeli A, Guglielmi G, Dovio A, Capelli G, de Feo D, Giannini S, et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: A cross-sectional outpatient study. Bone 2006;39(2):253–9.
- 10. van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000;39(12):1383–9.
- 11. Goodman SM, Springer B, Guyatt G, Abdel MP, Dasa V, George M, et al. 2017 American College of Rheumatology/American Association of Hip and Knee Surgeons Guideline for the Perioperative Management of Antirheumatic Medication in Patients With Rheumatic Diseases Undergoing Elective Total Hip or Total Knee Arthroplasty. Arthritis Care Res (Hoboken) 2017;69(8):1111–24.